refdb_folder <- here::here("data", "refdb")
refdb_folder[1] "/Users/marcgarel/Desktop/formation_metabarcoding_omic/data/refdb"To begin with, download the course repository on your computer or virtual machine.
To do so, visit the repository on github (ANF-metabarcoding) and download it as a zip file.
Once on your machine, unzip the file and place the resulting folder in the most covenient location (~/Documents/ for example).
Save as a variable the path to the folder where you will place the references databases.
refdb_folder <- here::here("data", "refdb")
refdb_folder[1] "/Users/marcgarel/Desktop/formation_metabarcoding_omic/data/refdb"The reason why we use here::here() is that when you render a Rmarkdown file, the working directory is where the Rmarkdown file is:
getwd()Whereas here::here() point to the root of the R project
here::here()[1] "/Users/marcgarel/Desktop/formation_metabarcoding_omic"Now, let’s create the folder directly from R:
if (!dir.exists(refdb_folder)) dir.create(refdb_folder)You can also create the folder from RStudio in the Files window
Tip: You can access the documentation of any R function using ? in the console. If you to know everything about the function dir.create(), simply run ?dir.create()
The Silva reference database, commonly used to assign 16S metabarcoding data, will be used in practical.
In case you are working with 18S sequences, you will have better assignements using PR2 (https://pr2-database.org/) or EukRibo (https://doi.org/10.5281/zenodo.6327891) especially if you are interested in protists.
The following code downloads dada2 formated silva reference databases. If you are not confortable with it, you can simply download the reference database files from your web browser here and here.
# R stop dowloading after timeout which is
# 60 seconds by default
getOption("timeout")[1] 60# so we change timeout to be 20 minutes
options(timeout = 1200)
# we save in variable the path to the refdb
# in the working space
silva_train_set <- file.path(refdb_folder,
"silva_nr99_v138.1_train_set.fa.gz")
silva_species_assignment <- file.path(refdb_folder,
"silva_species_assignment_v138.1.fa.gz")
# then we download the files if they don't already exist
if (!file.exists(silva_train_set)) {
download.file(
"https://zenodo.org/record/4587955/files/silva_nr99_v138.1_train_set.fa.gz",
silva_train_set,
quiet = TRUE
)
}
if (!file.exists(silva_species_assignment)) {
download.file(
"https://zenodo.org/record/4587955/files/silva_species_assignment_v138.1.fa.gz",
silva_species_assignment,
quiet = TRUE
)
}We will use in this practical R functions especially written for this course. The “classic” way to import functions is to use source() with the name of the R script to source.
source(here::here("scripts", "R", "preprocessing.R"))Save the path to the directory containing your raw data (paired-end sequencing fastq files) in a variable named path_to_fastqs
path_to_fastqs <- here::here("data", "raw")The gzipped (compressed) FASTQ formated “forward” (R1) and “reverse” (R2) files are named as follow:
${SAMPLENAME}_R1.fastq.gz for the forward files${SAMPLENAME}_R2.fastq.gz for the reverse files.We list the forward files using the function list.files(). The argument pattern gives you the possibility to only select file names matching a regular expression. In our case, we select all file names finising by _R1.fastq.gz.
fnFs <- sort(list.files(path_to_fastqs,
pattern = "_R1.fastq.gz",
full.names = TRUE))We do the same for reverse samples.
fnRs <- sort(list.files(path_to_fastqs,
pattern = "_R2.fastq.gz",
full.names = TRUE))To understand: What fnFs & fnRs variables contain?
head(fnFs)[1] "/Users/marcgarel/Desktop/formation_metabarcoding_omic/data/raw/S11B_R1.fastq.gz"
[2] "/Users/marcgarel/Desktop/formation_metabarcoding_omic/data/raw/S1B_R1.fastq.gz"
[3] "/Users/marcgarel/Desktop/formation_metabarcoding_omic/data/raw/S2B_R1.fastq.gz"
[4] "/Users/marcgarel/Desktop/formation_metabarcoding_omic/data/raw/S2S_R1.fastq.gz"
[5] "/Users/marcgarel/Desktop/formation_metabarcoding_omic/data/raw/S3B_R1.fastq.gz"
[6] "/Users/marcgarel/Desktop/formation_metabarcoding_omic/data/raw/S3S_R1.fastq.gz" sample_names <- basename(fnFs) |>
strsplit(split = "_") |>
sapply(`[`, 1)To understand:
basename(): remove path to only keep file name.
|>: R “pipe”. It allows you to chain functions, avoiding intermediate variables and nested parenthesis. It basically transfers the output of the left expression to the input of the right expression. You need R > 4.1 to use this pipe, otherwise use %>% from magrittr.
strsplit(): split character chain according to a defined pattern. ?strsplit for documentation.
sapply(): apply a function to each element of a list or vector. The output is simplified to be vector.
Let’s go step by step. First list the R1 file names.
basename(fnFs) |>
head()[1] "S11B_R1.fastq.gz" "S1B_R1.fastq.gz" "S2B_R1.fastq.gz" "S2S_R1.fastq.gz"
[5] "S3B_R1.fastq.gz" "S3S_R1.fastq.gz" We can see that the sample name is before the first _. With strsplit(), we can split each file name into a 2 elements vector. The result is a list of 2 elements vectors.
basename(fnFs) |>
strsplit(split = "_") |>
head()[[1]]
[1] "S11B" "R1.fastq.gz"
[[2]]
[1] "S1B" "R1.fastq.gz"
[[3]]
[1] "S2B" "R1.fastq.gz"
[[4]]
[1] "S2S" "R1.fastq.gz"
[[5]]
[1] "S3B" "R1.fastq.gz"
[[6]]
[1] "S3S" "R1.fastq.gz"Now, We just have to extract the first element for each file.
basename(fnFs) |>
strsplit(split = "_") |>
sapply(`[`, 1) |>
head()[1] "S11B" "S1B" "S2B" "S2S" "S3B" "S3S" Regular expressions are extremly useful. If you are keen to learn how to use them, have a look here
We use a custom function, qualityprofile(), implemented in R/preprocessing.R to check the quality of the raw sequences.
Run ?qualityprofile to know more about this function.
# create a directory for the outputs
quality_folder <- here::here("outputs",
"dada2",
"quality_plots")
if (!dir.exists(quality_folder)) {
dir.create(quality_folder, recursive = TRUE)
}
qualityprofile(fnFs,
fnRs,
file.path(quality_folder, "quality_plots.pdf"))Open the the pdf file generated by qualityprofile()
We first create a folder where to save the reads once they are trimmed:
path_to_trimmed_reads <- here::here("outputs","dada2", "trimmed")
if (!dir.exists(path_to_trimmed_reads)) dir.create(path_to_trimmed_reads)Then we prepare a list of paths where to save the results (e.g. sequences without primers)
nopFw <- file.path(path_to_trimmed_reads, basename(fnFs))
nopRv <- file.path(path_to_trimmed_reads, basename(fnRs))
head(nopFw)[1] "/Users/marcgarel/Desktop/formation_metabarcoding_omic/outputs/dada2/trimmed/S11B_R1.fastq.gz"
[2] "/Users/marcgarel/Desktop/formation_metabarcoding_omic/outputs/dada2/trimmed/S1B_R1.fastq.gz"
[3] "/Users/marcgarel/Desktop/formation_metabarcoding_omic/outputs/dada2/trimmed/S2B_R1.fastq.gz"
[4] "/Users/marcgarel/Desktop/formation_metabarcoding_omic/outputs/dada2/trimmed/S2S_R1.fastq.gz"
[5] "/Users/marcgarel/Desktop/formation_metabarcoding_omic/outputs/dada2/trimmed/S3B_R1.fastq.gz"
[6] "/Users/marcgarel/Desktop/formation_metabarcoding_omic/outputs/dada2/trimmed/S3S_R1.fastq.gz" The data you are working with correspond to the V3-V4 region using the primers Pro341F (CCTACGGGNBGCASCAG) and Pro805R (GACTACNVGGGTATCTAAT) Save into variables the forward and reverse primers.
primer_fwd <- "CCTACGGGNBGCASCAG"
#regular expression: CCTACGGG.[CGT]GCA[GC]CAG
primer_rev <- "GACTACNVGGGTATCTAAT"dada2::removePrimers(fn = fnFs,
fout = nopFw,
primer.fwd = primer_fwd,
max.mismatch = 1,
verbose = TRUE)
dada2::removePrimers(fn = fnRs,
fout = nopRv,
primer.fwd = primer_rev,
max.mismatch = 1,
verbose = TRUE)Same as before, create a folder
path_to_filtered_reads <- here::here("outputs", "dada2", "filtered")
if (!dir.exists(path_to_filtered_reads)) dir.create(path_to_filtered_reads)and list paths:
filtFs <- file.path(path_to_filtered_reads, basename(fnFs))
filtRs <- file.path(path_to_filtered_reads, basename(fnRs))To make the link between files and sample names, simply name the file names vector using the sample names
names(filtFs) <- sample_names
names(filtRs) <- sample_names
names(nopFw) <- sample_names
names(nopRv) <- sample_namesdada2::filterAndTrim()Let’s have a look at what the function is doing. To do so, type ?dada2::filterAndTrim() in the console.
Let’s run the function.
(out <- dada2::filterAndTrim(
fwd = nopFw,
filt = filtFs,
rev = nopRv,
filt.rev = filtRs,
minLen = 150,
matchIDs = TRUE,
maxN = 0,
maxEE = c(3, 3),
truncQ = 2
)) reads.in reads.out
S11B_R1.fastq.gz 1985 994
S1B_R1.fastq.gz 1973 1051
S2B_R1.fastq.gz 1985 1058
S2S_R1.fastq.gz 1981 1046
S3B_R1.fastq.gz 1986 1044
S3S_R1.fastq.gz 1987 1132
S4B_R1.fastq.gz 1980 1058
S4S_R1.fastq.gz 1983 1135
S5B_R1.fastq.gz 1984 1049
S5S_R1.fastq.gz 1988 1055
S6B_R1.fastq.gz 1979 1035
S6S_R1.fastq.gz 1979 1025
S7B_R1.fastq.gz 1985 997
S7S_R1.fastq.gz 1985 1008
S8B_R1.fastq.gz 1992 976
S8S_R1.fastq.gz 1980 1051
S9B_R1.fastq.gz 1979 956
S9S_R1.fastq.gz 1979 1045What happened?
Details about the function arguments: * nopFw : input, where the forward reads without primers are (path) * filtFs : output, where forward filtered reads are written (path) * nopRv and filRs : same as above, but wiht reverse reads * TruncLen : truncate reads after truncLen bases. Reads shorter than that are discarded. Exple : TruncLen=c(200,150), means forward and reverse reads are cut at 200 bp and 150 bp respectively. * TrimLeft : number of nucleotides to remove from the start * Trimright : number of nucleotides to remove from the end * maxN : max number of ambiguous bases accepted * maxEE : read expected errors (EE) threshold. The EE of a read is the sum of the error probability of each base composing it. Increase that value to accept more low quality reads. The first value refers to the forward reads and the second to the reverse reads. * TruncQ=2: truncate reads at the first instance of a quality score less than or equal to truncQ.
To be able to denoise your data, you need an error model. The error model will tell you at which rate a nucleotide is replace by another for a given quality score. For example, for a quality score Q of 30, what is the probability of an A being wrongly read as a T.
This error model can be learnt directly from the data with the function dada2::learnErrors(). You can come back to the course for more details about the maths behind.
errF <- dada2::learnErrors(filtFs,
randomize = TRUE,
multithread = TRUE)5155290 total bases in 18715 reads from 18 samples will be used for learning the error rates.errR <- dada2::learnErrors(filtRs,
randomize = TRUE,
multithread = TRUE)5277647 total bases in 18715 reads from 18 samples will be used for learning the error rates.You can visualise the resulting error model using the function dada2::plotErrors()
dada2::plotErrors(errF, nominalQ=TRUE)Warning in scale_y_log10(): log-10 transformation introduced infinite values.
log-10 transformation introduced infinite values.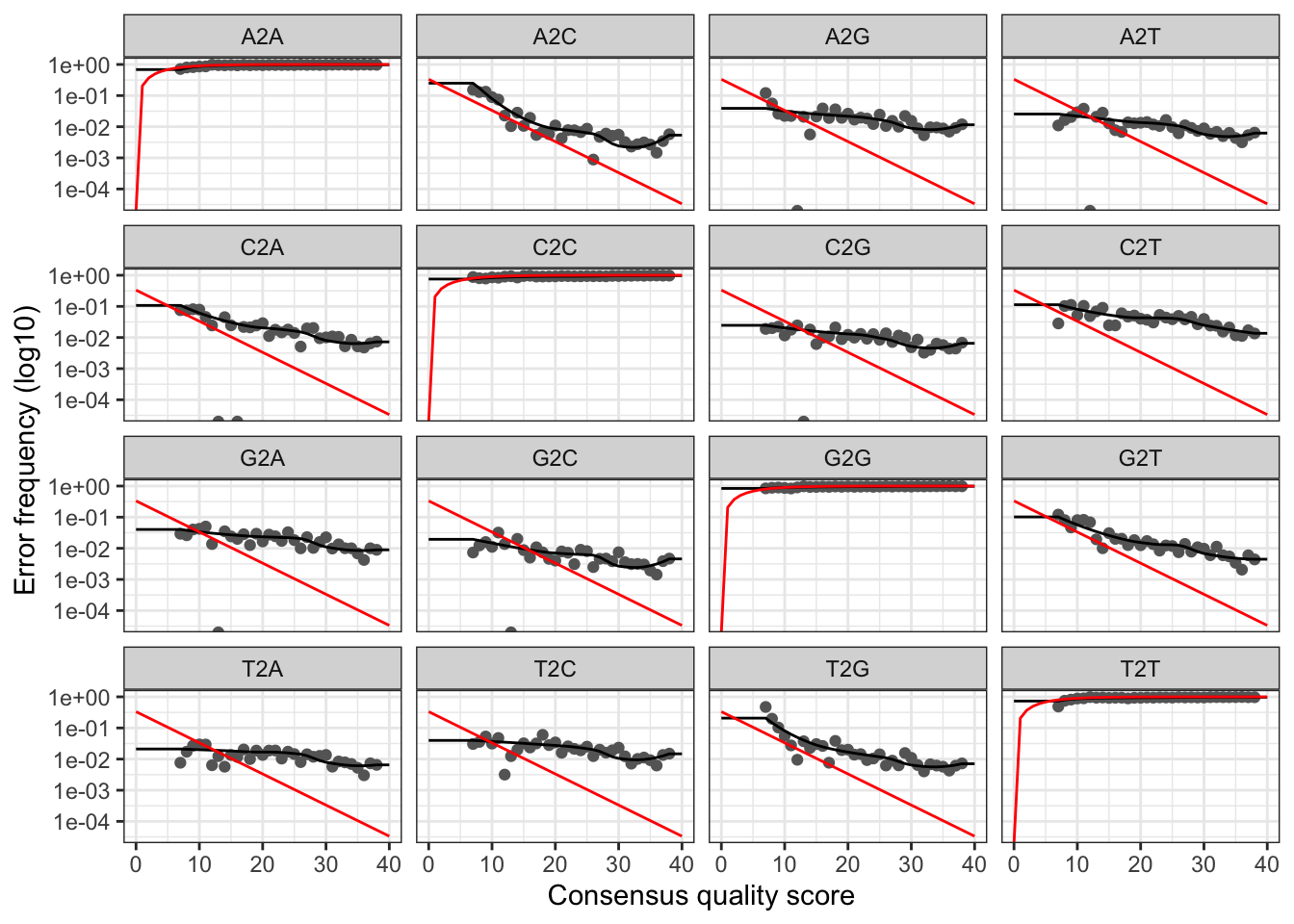
Before denoising, we need to dereplicate the sequences. It means, for each unique sequence, count the number of reads.
The dereplication is achieved using the function dada2::derepFastq()
derepFs <- dada2::derepFastq(filtFs, verbose = TRUE)
derepRs <- dada2::derepFastq(filtRs, verbose = TRUE)Now we are ready to run the denoising algorithm with dada2::dada(). As input, we need the error model and the dereplicated sequences.
dadaFs <- dada2::dada(derepFs, err = errF, multithread = TRUE)Sample 1 - 994 reads in 633 unique sequences.
Sample 2 - 1051 reads in 683 unique sequences.
Sample 3 - 1058 reads in 671 unique sequences.
Sample 4 - 1046 reads in 661 unique sequences.
Sample 5 - 1044 reads in 670 unique sequences.
Sample 6 - 1132 reads in 665 unique sequences.
Sample 7 - 1058 reads in 649 unique sequences.
Sample 8 - 1135 reads in 563 unique sequences.
Sample 9 - 1049 reads in 668 unique sequences.
Sample 10 - 1055 reads in 578 unique sequences.
Sample 11 - 1035 reads in 597 unique sequences.
Sample 12 - 1025 reads in 563 unique sequences.
Sample 13 - 997 reads in 586 unique sequences.
Sample 14 - 1008 reads in 578 unique sequences.
Sample 15 - 976 reads in 595 unique sequences.
Sample 16 - 1051 reads in 604 unique sequences.
Sample 17 - 956 reads in 566 unique sequences.
Sample 18 - 1045 reads in 582 unique sequences.dadaRs <- dada2::dada(derepRs, err = errR, multithread = TRUE)Sample 1 - 994 reads in 730 unique sequences.
Sample 2 - 1051 reads in 763 unique sequences.
Sample 3 - 1058 reads in 780 unique sequences.
Sample 4 - 1046 reads in 747 unique sequences.
Sample 5 - 1044 reads in 762 unique sequences.
Sample 6 - 1132 reads in 796 unique sequences.
Sample 7 - 1058 reads in 763 unique sequences.
Sample 8 - 1135 reads in 733 unique sequences.
Sample 9 - 1049 reads in 777 unique sequences.
Sample 10 - 1055 reads in 718 unique sequences.
Sample 11 - 1035 reads in 727 unique sequences.
Sample 12 - 1025 reads in 672 unique sequences.
Sample 13 - 997 reads in 700 unique sequences.
Sample 14 - 1008 reads in 709 unique sequences.
Sample 15 - 976 reads in 699 unique sequences.
Sample 16 - 1051 reads in 762 unique sequences.
Sample 17 - 956 reads in 674 unique sequences.
Sample 18 - 1045 reads in 724 unique sequences.Once forward and reverse reads have been denoised, we can merge them with dada2::mergePairs().
mergers <- dada2::mergePairs(
dadaF = dadaFs,
derepF = derepFs,
dadaR = dadaRs,
derepR = derepRs,
maxMismatch = 0,
verbose = TRUE
)At this point we have ASVs and we know their number of reads in each sample. Whe have enough information to build an ASV table.
seqtab <- dada2::makeSequenceTable(mergers)Chimeras are artifact sequences formed by two or more biological sequences incorrectly joined together. We find and remove bimeras (two-parent chimeras) using the function dada2::removeBimeraDenovo()
seqtab_nochim <- dada2::removeBimeraDenovo(seqtab,
method = "consensus",
multithread = TRUE,
verbose = TRUE)Statistics about each preprocessing can also be exported. First this table need to be assembled:
getN <- function(x) sum(dada2::getUniques(x))
summarytab <- data.frame(Samples=sample_names,
imput=out[,1],
filtered=out[,2],
denoised=sapply(dadaFs,getN),
merged=sapply(mergers,getN),
nonchimeric=rowSums(seqtab_nochim),
Final_retained=rowSums(seqtab_nochim)/out[,1]*100)
summarytab Samples imput filtered denoised merged nonchimeric
S11B_R1.fastq.gz S11B 1985 994 884 749 749
S1B_R1.fastq.gz S1B 1973 1051 823 708 708
S2B_R1.fastq.gz S2B 1985 1058 882 672 672
S2S_R1.fastq.gz S2S 1981 1046 917 783 783
S3B_R1.fastq.gz S3B 1986 1044 857 672 672
S3S_R1.fastq.gz S3S 1987 1132 940 776 776
S4B_R1.fastq.gz S4B 1980 1058 887 687 687
S4S_R1.fastq.gz S4S 1983 1135 967 870 870
S5B_R1.fastq.gz S5B 1984 1049 933 770 770
S5S_R1.fastq.gz S5S 1988 1055 892 767 766
S6B_R1.fastq.gz S6B 1979 1035 948 817 817
S6S_R1.fastq.gz S6S 1979 1025 870 738 738
S7B_R1.fastq.gz S7B 1985 997 868 684 684
S7S_R1.fastq.gz S7S 1985 1008 874 754 754
S8B_R1.fastq.gz S8B 1992 976 811 694 694
S8S_R1.fastq.gz S8S 1980 1051 903 742 742
S9B_R1.fastq.gz S9B 1979 956 840 650 650
S9S_R1.fastq.gz S9S 1979 1045 904 746 746
Final_retained
S11B_R1.fastq.gz 37.73300
S1B_R1.fastq.gz 35.88444
S2B_R1.fastq.gz 33.85390
S2S_R1.fastq.gz 39.52549
S3B_R1.fastq.gz 33.83686
S3S_R1.fastq.gz 39.05385
S4B_R1.fastq.gz 34.69697
S4S_R1.fastq.gz 43.87292
S5B_R1.fastq.gz 38.81048
S5S_R1.fastq.gz 38.53119
S6B_R1.fastq.gz 41.28348
S6S_R1.fastq.gz 37.29156
S7B_R1.fastq.gz 34.45844
S7S_R1.fastq.gz 37.98489
S8B_R1.fastq.gz 34.83936
S8S_R1.fastq.gz 37.47475
S9B_R1.fastq.gz 32.84487
S9S_R1.fastq.gz 37.69581The ASV table is ready. But without a clue about the ASVs taxonomic identity, we won’t go far in ecological interpretations. We can have an idea of ASV taxonomic identity comparing their sequences to references databases such as SILVA.
The taxonomic assignment is done in two steps.
First, each ASV is assigned to a taxonomy using the RDP Naive Bayesian Classifier algorithm described in Wang et al. 2007 called by the function dada2::assignTaxonomy().
taxonomy <- dada2::assignTaxonomy(
seqs = seqtab_nochim,
refFasta = silva_train_set,
taxLevels = c("Kingdom", "Phylum", "Class",
"Order", "Family", "Genus",
"Species"),
multithread = TRUE,
minBoot = 60
)The method is robust, however, it often fails to assign at the species level.
If you consider that in case an ASV is 100% similar to a reference sequence, it belongs to the same species, then you can use dada2::addSpecies()
taxonomy <- dada2::addSpecies(taxonomy,
silva_species_assignment,
allowMultiple = FALSE)This function assign to the species level ASVs which are identical to a reference sequence.
We will also need some information about the samples, the file is located is another folder.
context <- read.table(here::here("data",
"mapfileFA.txt"),
header = TRUE,row.names = 1)
#See
context SampName Geo Description groupe Pres PicoEuk Synec Prochloro NanoEuk
S1B S1B North North1B NBF 52 660 32195 10675 955
S2B S2B North North2B NBF 59 890 25480 16595 670
S2S S2S North North2S NBS 0 890 25480 16595 670
S3B S3B North North3B NBF 74 835 13340 25115 1115
S3S S3S North North3S NBS 0 715 26725 16860 890
S4B S4B North North4B NBF 78 2220 3130 29835 2120
S4S S4S North North4S NBS 78 2220 3130 29835 2120
S5B S5B North North5B NBF 42 1620 55780 23795 2555
S5S S5S North North5S NBS 0 1620 56555 22835 2560
S6B S6B South South1B SGF 13 2520 39050 705 3630
S6S S6S South South1S SGS 0 2435 35890 915 3735
S7B S7B South South2B SGF 26 0 0 0 4005
S7S S7S South South2S SGS 0 4535 26545 1340 6585
S8B S8B South South3B SGF 33 0 0 0 5910
S8S S8S South South3S SGS 0 4260 36745 985 5470
S9B S9B South South4B SGF 25 4000 31730 485 4395
S9S S9S South South4S SGS 0 5465 32860 820 5045
S11B S11B South South5B SGF 35 5370 46830 580 6010
Crypto SiOH4 NO2 NO3 NH4 PO4 NT PT Chla T S
S1B 115 1.813 0.256 0.889 0.324 0.132 9.946 3.565 0.0000 22.7338 37.6204
S2B 395 2.592 0.105 1.125 0.328 0.067 9.378 3.391 0.0000 22.6824 37.6627
S2S 395 3.381 0.231 0.706 0.450 0.109 8.817 3.345 0.0000 22.6854 37.6176
S3B 165 1.438 0.057 1.159 0.369 0.174 8.989 2.568 0.0000 21.5296 37.5549
S3S 200 1.656 0.098 0.794 0.367 0.095 7.847 2.520 0.0000 22.5610 37.5960
S4B 235 2.457 0.099 1.087 0.349 0.137 8.689 3.129 0.0000 18.8515 37.4542
S4S 235 2.457 0.099 1.087 0.349 0.137 8.689 3.129 0.0000 18.8515 37.4542
S5B 1355 2.028 0.103 1.135 0.216 0.128 8.623 3.137 0.0102 24.1905 38.3192
S5S 945 2.669 0.136 0.785 0.267 0.114 9.146 3.062 0.0000 24.1789 38.3213
S6B 1295 2.206 0.249 0.768 0.629 0.236 9.013 3.455 0.0000 22.0197 39.0877
S6S 1300 3.004 0.251 0.927 0.653 0.266 8.776 3.230 0.0134 22.0515 39.0884
S7B 1600 3.016 0.157 0.895 0.491 0.176 8.968 4.116 0.0000 23.6669 38.9699
S7S 1355 1.198 0.165 1.099 0.432 0.180 8.256 3.182 0.0000 23.6814 38.9708
S8B 1590 3.868 0.253 0.567 0.533 0.169 8.395 3.126 0.0000 23.1236 39.0054
S8S 2265 3.639 0.255 0.658 0.665 0.247 8.991 3.843 0.0132 23.3147 38.9885
S9B 1180 3.910 0.107 0.672 0.490 0.134 8.954 4.042 0.0172 22.6306 38.9094
S9S 1545 3.607 0.139 0.644 0.373 0.167 9.817 3.689 0.0062 22.9545 38.7777
S11B 1690 3.324 0.083 0.756 0.467 0.115 9.539 4.138 0.0182 23.0308 38.9967
Sigma_t
S1B 26.0046
S2B 26.0521
S2S 26.0137
S3B 26.2987
S3S 26.0332
S4B 26.9415
S4S 26.9415
S5B 26.1037
S5S 26.1065
S6B 27.3241
S6S 27.3151
S7B 26.7536
S7S 26.7488
S8B 26.9423
S8S 26.8713
S9B 27.0131
S9S 26.8172
S11B 26.9631physeq <- phyloseq::phyloseq(
phyloseq::otu_table(seqtab_nochim,taxa_are_rows = FALSE),
phyloseq::tax_table(taxonomy),
phyloseq::sample_data(context)
)
#See
physeqphyloseq-class experiment-level object
otu_table() OTU Table: [ 155 taxa and 18 samples ]
sample_data() Sample Data: [ 18 samples by 21 sample variables ]
tax_table() Taxonomy Table: [ 155 taxa by 7 taxonomic ranks ]The function will add sequences AND change labels automatically with ASV1,ASV2 etc
physeq <-microbiome::add_refseq(physeq)
#See
physeqphyloseq-class experiment-level object
otu_table() OTU Table: [ 155 taxa and 18 samples ]
sample_data() Sample Data: [ 18 samples by 21 sample variables ]
tax_table() Taxonomy Table: [ 155 taxa by 7 taxonomic ranks ]
refseq() DNAStringSet: [ 155 reference sequences ]Knowing the ASVs phylogenetic relatedness will help you to have a better understanding of the communities your studying.
To quote the evolutionary biologist Theodosius Dobzhansky:
Nothing in Biology Makes Sense Except in the Light of Evolution.
A phylogenetic tree reconstructed from the ASV sequences will be used to measure their relatedness.
Before reconstructing a phylogenetic tree we need to align the ASVs sequences.
aln <- DECIPHER::AlignSeqs(phyloseq::refseq(physeq), anchor = NA)Once it is done, you can visualise the alignment using the function DECIPHER::BrowseSeqs()
DECIPHER::BrowseSeqs(aln, highlight = 0)We will infer a phylogenetic from our alignement using the library phangorn.
First, let’s convert our DNAStringSet alignment to the phangorn phyDat format.
phang_align <- as.matrix(aln) |> phangorn::phyDat(type = "DNA")Then, we compute pairwise distances of our aligned sequences using equal base frequencies (JC69 model used by default).
dm <- phangorn::dist.ml(phang_align, model = "JC69")Finally, we reconstruct a neighbour joining tree.
treeNJ <- phangorn::NJ(dm)Other approaches to reconstruct a phylogenetic tree exist. If you want to try them with phangorn, have a look here
We need the tree to be rooted for future analysis. We can do that using the function phangorn::midpoint()
Change label (leaves) of the tree (important)
treeNJ <- phangorn::midpoint(tree = treeNJ)Once we have a rooted tree, we can add it to the phyloseq object.
physeq <- phyloseq::merge_phyloseq(physeq,treeNJ)
#See
physeqphyloseq-class experiment-level object
otu_table() OTU Table: [ 155 taxa and 18 samples ]
sample_data() Sample Data: [ 18 samples by 21 sample variables ]
tax_table() Taxonomy Table: [ 155 taxa by 7 taxonomic ranks ]
phy_tree() Phylogenetic Tree: [ 155 tips and 154 internal nodes ]
refseq() DNAStringSet: [ 155 reference sequences ]directory_output_phyloseq <- here::here("outputs","dada2", "Phyloseq")
if (!dir.exists(directory_output_phyloseq)) dir.create(directory_output_phyloseq)
phylobject <- file.path(directory_output_phyloseq,"myphylobject.RData")save(physeq,file=phylobject)